The MastAttack 107: The Layperson’s Guide to Understanding Mast Cell Diseases, Part 67
81. Is it true almost 20% of the population might have mast cell disease?
- We need large scale studies in order to know for sure but this is very unlikely.
- This figure first appeared in a paper published in 2013. This study asked mast cell patients, their relatives, and healthy control subjects to complete a questionnaire about whether or not they had symptoms of mast cell disease. 17% of the patients who were in the healthy control group reported symptoms that mast cell patients may experience. However, as we all know, you can have symptoms for a lot of reasons. Just sharing symptoms doesn’t mean anything. Also, surveys and questionnaires are known to be a bad way to collect hard data. By nature, they ask leading questions and are extremely subjective.
- This same paper also tested for CKIT mutations in 20 mast cell patients and 20 healthy controls. The mast cell patient group included patients with MCAS, systemic mastocytosis, and cutaneous mastocytosis. The paper reported that 13/20 (65%) mast cell patients had mutations in the CKIT gene while 3/20 healthy controls (15%) had mutations in the CKIT gene. However, these mutations are not known to cause mast cell disease, or any diseases, for that matter. 20 people is a TINY number for a genetics study. You need a much larger group to really know whether or not a mutation occurs in healthy people or just people with a specific disease. We are talking hundreds to thousands of people needed in order to really know.
- Many mastocytosis patients have a specific mutation called the CKIT D816V mutation. This mutation in mast cells only occurs when the patient has a clonal mast cell disorder like mastocytosis. People in the general population do not have the CKIT D816V without having mast cell disease.
- Currently, the only CKIT mutations known to be associated with mast cell disease are the mutations at codon 816 of the CKIT gene. The D816V mutation is overwhelmingly the most common, but there are others that same exact spot, including D816Y, and a few others. Other mutations in the CKIT mutation are not known to be associated with mast cell disease.
- Genes mostly tell your cells how to make proteins. The reason mutations can cause diseases is because they can change the structure of the protein that is made, affecting how it works. But not all mutations cause disease. Many mutations don’t change the proteins made by genes. This means that just having a mutation does not necessarily mean you have a disease. Mutations have to be linked to a disease by experimental work showing that people with a particular mutation have a particular disease.
- Mutations are really very common. They occur so frequently that your cells have lots of failsafes in place specifically to work around or fix mutations. There are several ways they can do this but I’m just going to talk about one right now.
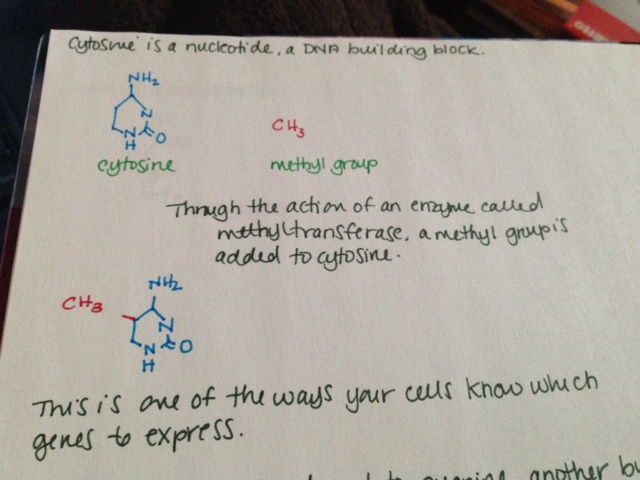
- As I mentioned above, genes tell your cells how to make proteins. Genes are made of DNA. DNA is made of tiny building blocks called nucleotides. There are four kinds of DNA building blocks. These building blocks are grouped in bundles of three. Every one of those three DNA building block bundles tells the cell how to make a little piece of the protein. Those bundles are called codons. Then the three DNA building blocks next to that bundle tell the cell how to make another little piece of the protein, and so on. Those little pieces stick together and make the full sized protein.
- The cell is able to make a protein by reading through this gene three pieces at a time. This is how our cells use genes to make proteins.
- So now we know that those three DNA building blocks work together to me a tiny piece of protein. There are four kinds of DNA building blocks. What piece of protein is made is determined by which three building blocks are grouped together. There are many combinations of building blocks.
- If one of those building blocks is mutated, it could cause the wrong protein piece to be made. So it seems that each of the three building blocks is really important to making the right protein. However, this is not the case. In fact, of those three building blocks, the last one is mostly irrelevant. If it’s mutated, it will usually still make the right protein piece. In some cases, the second one isn’t important either. The first one is the most important, but having a mutation there doesn’t always mean the protein is made wrong either.
- So genes could potentially have up to 1/3 of its building blocks mutated without causing a problem. (I’m being very general here.) This phenomenon, in which the third DNA building block in a group can be wrong without messing up the protein is called wobble. Wobble is a built in mechanism that allows cells to make mutations sometimes without consequences.
- That’s a lot of mutations that don’t cause problems. That’s a lot of mutations that still allow the cell to make the right protein. That’s a lot of mutations without causing symptoms or disease.
- Your genes can withstand a lot of changes to single building blocks. When a single nucleotide is changed, it is called a single nucleotide polymorphism (snp). Most of these snps don’t cause trouble at all. The only way to know which ones cause problems is to gather up a bunch of people with these mutations and study them to see if they have diseases and, if so, which ones. So just having a mutation is not enough to know if it causes diseases without further study.
For additional reading, please visit the following posts:
Gene expression and the CKIT D816V mutation
The MastAttack 107: The Layperson’s Guide to Understanding Mast Cell Diseases, Part 2
The difference between C117+ and CKIT+
Heritable mutations in mastocytosis