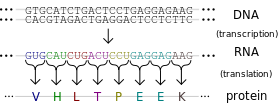
MTHFR (methylenetetrahydrofolate reductase) is an enzyme involved in folate metabolism. It is rate limiting, which means that if there is not enough, your body cannot metabolize enough folate; if there is too much, it metabolizes too much. How much folate your body is able to metabolize is directly related to how much MTHFR you have in your body. Some folate broken down by your body is used to methylate DNA. There has been much conflicting evidence, but it seems that low folate in the body is associated with less DNA methylation overall, which may be associated with cancer (Crider, 2012.)
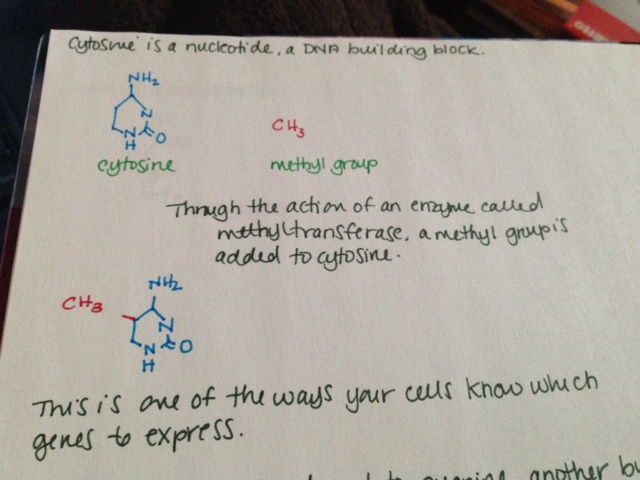
A single nucleotide polymorphism (SNP) is a change in DNA sequence in which one nucleotide (a DNA building block) is changed. Importantly, SNPs are common. In fact, they are so common that the way your body codes its DNA allows for SNPs to not change gene expression in many cases. This is called wobble. Three DNA building blocks in a row make one amino acid, which is used to build proteins that do things in the body. However, in many cases, the third building block can be any building block and it will still make the same amino acid. Please consider this. SNPs are so common that your cells know that in many cases, 1 in every 3 DNA building blocks can be anything and it won’t change a thing.
SNPs have become the topic of increased interest, both by medical and scientific professions and by lay people. It is certainly true that some SNPs play a role in development and progression of diseases. MTHFR has been found to have up to 24 reported SNPs, with two being of particular interest. These are C677T and A1298C.
The normal (or “wildtype”) form of MTHFR has a cytosine (C) nucleotide where people with the C677T mutation have a thymine nucleotide. If you have two copies of the regular gene, you are 677CC and homozygous for the regular form of the gene (the allele.) If you are 677CT, you have one copy of the allele with the SNP. If you are 677TT, you have two copies of the allele with the SNP. About 10% of North Americans are 677TT, meaning they have two copies of the mutated allele. It is most common in Hispanics and those of Mediterranean descent, next most common in Caucasians and least common in African Americans (Schneider, 1998.)
Being homozygous for 677TT can cause a mild MTHFR deficiency because that this form of the enzyme is generally less stable, which means it breaks down faster than usual. People with this profile are also more likely to develop mild hyperhomocysteinemia, an elevation of homocysteine in the blood. Homocysteine is consumed in the metabolism of folate, and because there is less MTHFR with the 677TT mutation, the homocysteine is not all getting used. It is also elevated in cases of B6, B12 and folic acid deficiency.
Increased homocysteine has been studied for its possible relationship to health issues, including increased clotting, strokes, schizophrenia and osteoporosis. Despite multiple studies (to be honest, quite a lot of studies), the results are really non-uniform. Because it was linked earlier to cardiovascular disease, multiple studies investigated the benefits of lowering homocysteine. In diabetic nephropathy patients, treating to lower homocysteine actually doubled cardiovascular events including heart attack and stroke, some leading to death, as well as decreased renal function (House, 2010.) But another study found lowering homocysteine decreased the risk of stroke by as much as 25% (Lonn, 2006.)
The effects of the C677T SNP have been most well studied in regards to development of cancers. Folic acid is a key metabolite in the development and proliferation of cells, so deficiency can be limiting to cell division. 677TT patients who are not folate deficient are actually 50% less likely to develop colorectal cancer or colorectal adenoma (Chen, 1999.) 677TT patients who are folate deficient have at least the same risk as 677CC patients and potentially more risk (Slattery, 1999.) 677TT patients are over four times less likely to develop acute lymphocytic leukemia. They have the same risk of developing acute myeloid leukemia (Skibola, 1999.) Some studies found them to have increased risk of cervical neoplasia, breast cancer, endometrial cancer and gastric cancer, but all of these studies were done on small populations (Xia, 2014.)
The other MTHFR SNP that gets a lot of attention is A1298C. At position 1298 of the MTHFR gene, the wildtype allele has adenine. In some people, it is substituted for cytosine. Wildtype is homozygous for adenine there and they are called 1298AA. If you have one copy of the SNP, you are 1298AC. If you have two copies, you are 1298CC. The A1298C has much less effect on the stability of the MTHFR protein than the C677T mutation. It is not known to cause elevation of homocysteine levels. There has been a lot of controversy over whether this mutation can cause a deficiency of BH4, tetrahydrobiopterin. BH4 is important in formation of neurotransmitters and nitric oxide, as well as consuming ammonia. Low levels of BH4 have been tied to phenylketonuria and trials with BH4 supplementation have seen encouraging results (Michals-Matalon, 2007.)
In the last six months or so, I have read about fifty scientific papers on MTHFR or related topics. I did this because I originally planned an MTHFR post for last summer. I didn’t do a post because the data is a mess. You cannot ascertain much of use from the peer reviewed literature on the C677T and A1298C mutations – and not for lack of effort. These mutations have been very well studied.
MTHFR mutations and methylation are talked about a lot in the mast cell community. Many people believe that having an MTHFR mutation severely impacts folate metabolism, which in turn means there is not enough methylation, and this dysregulation causes overexpression of genes causing disease. I have searched thoroughly for a link. Really thoroughly. I cannot find any link that is not the idea of one person and researched by that one person, usually outside of peer reviewed settings. I cannot find any link that is not described in detail by a person who does not stand to gain financially from patients who share their beliefs. I am not saying that MTHFR is definitely not linked to mast cell disease. I’m saying I can’t find any proof that it is. I can’t even find anything that SUGGESTS that it is. Might people with these mutations feel better with appropriate folic acid supplementation? Probably. Is that the same thing as causing mast cell disease? Certainly not. It is certainly not the same thing.
I think personal stories hold a lot of power. If your personal story is that your mast cell disease is significantly better controlled by addressing your MTHFR mutation, then I think that is fantastic. I think it is entirely possible that this is the case for many. But I do not believe it causes mast cell disease. And I think everyone would feel better with appropriate levels of folate, as it is an important player in many vital reactions.
References:
Schneider JA, Rees DC, Liu YT, Clegg JB (May 1998). “Worldwide distribution of a common methylenetetrahydrofolate reductase mutation”. Am. J. Hum. Genet. 62 (5): 1258–60
M L Slattery, et al. Methylenetetrahydrofolate reductase, diet and risk of colon cancer. Cancer Epidemiol Biomarkers Prev., 8 (1999), PP 560S-564S.
House, AA; Eliasziw, M; Cattran, DC; Churchill, DN; Oliver, MJ; Fine, A; Dresser, GK; Spence, JD (Apr 28, 2010). “Effect of B-vitamin therapy on progression of diabetic nephropathy: a randomized controlled trial.”. JAMA: the Journal of the American Medical Association 303 (16): 1603–9.
Lonn, E; Yusuf, S; Arnold, MJ; Sheridan, P; Pogue, J; Micks, M; McQueen, MJ; Probstfield, J; Fodor, G; Held, C; Genest J, Jr; Heart Outcomes Prevention Evaluation (HOPE) 2, Investigators (Apr 13, 2006). “Homocysteine lowering with folic acid and B vitamins in vascular disease.”. The New England Journal of Medicine 354 (15): 1567–77
Skibola CF, Smith MT, Kane E, Roman E, Rollinson S, Cartwright RA, Morgan G’ (October 1999). “Polymorphisms in the methylenetetrahydrofolate reductase gene are associated with susceptibility to acute leukemia in adults”. Proc. Natl. Acad. Sci. U.S.A. 96 (22): 12810–5.
Kim, Y I, et al. Methylenetetrahydrofolate reductase polymorphisms, folate and cancer risk: a paradigm of gene-nutrient interactions in carcinogenesis. Nutr Rev 58 (2000), pp 205-209.
Crider, Krista, et al. Folate and DNA Methylation: A Review of Molecular Mechanisms and the Evidence for Folate’s Role. Adv Nutr January 2012 Adv Nutr vol. 3: 21-38, 2012
Xia, Lei-Zhou, et al. Methylenetetrahydrofolate reductase C677T and A1298C polymorphisms and gastric cancer susceptibility. World J Gastroenterol. Aug 28, 2014; 20(32): 11429–11438.
J Chen, et al. MTHFR polymorphism, methyl-replete diets and the risk of colorectal carcinoma and adenoma among US men and women: an example of gene–environment interactions in colorectal tumorigenesis. J. Nutr., 129 (1999), pp. 560S–564S