Mast cell mutations: TET2 and mutation profiles of aggressive subtypes
TET2 (Tet methylcytosine dioxygenase 2) is found to be mutated in 20.8-29% of SM patients. Of note, dozens of mutations have been identified in this gene, including missense, nonsense, frameshift and deletion mutations. These mutations cause formation of a defective and less active TET2 enzyme. TET2 is located at chromosome 4q24 and mutations at this location are associated in both MPN and MDS conditions.
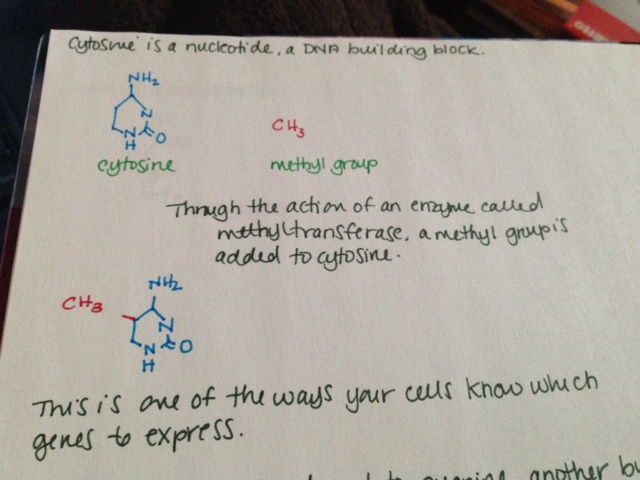
TET2 is involved in DNA methylation and demethylation, although the exact nature of this involvement is not clear. When a methyl group is added to a cytosine at a specific place in front of a gene, the gene is turned off and is not expressed. This is called “methylation.” TET2 adds a hydroxyl group to 5-methylcytosine, but it is not well understood if this turns the gene off. TET2 may also be involved in demethylating DNA, or removing those specific methyl groups. It has been shown to be involved with DNA demethylation during bone development.
One study looked at the mutational profiles of patients with various forms of SM, including ISM, SSM, SM-AHNMD, ASM and MCL, all of whom were positive for CKIT D816V mutation. 15/39 had a TET2 mutation. None of those patients had ISM or SSM. Of those with an aggressive form and a TET2 mutation, 67% had more than one TET2 mutation.
In this study, 24/27 patients with advanced SM (SM-AHNMD, ASM, MCL) had mutations beyond the D816V mutation. 5/5 SM-AHNMD patients and 19/22 ASM or MCL patients had multiple mutations (CKIT and something else.) In contrast, only 3/12 ISM or SSM patients had additional mutations. In advanced SM, 78% had at least 3 mutations, and 41% had at least 5.
These mutational profiles have clear implications clinically. 96% patients with major blood abnormalities (anemia <10 g/dL and/or thrombocytopenia < 100 x 10e9/L in addition to monocytosis > 1 x 10e9/L and/or eosinophilia >10%) had at least one additional molecular mutation regardless of SM subtype.
Advanced SM patients in this study all had one of the following multiple mutation profiles: 26% KIT-TET2-SRSF2, 18% KIT-SRSF2-RUNX1, 13% KIT-TET2-CBL, 10% KIT-SRSF2-ASXL1 10%, and 10% KIT-TET2-ASXL1. Patients with advanced SM (and therefore multiple mutations) were also found to be significantly older (68 years of age on average) than those with just the CKIT mutation (48 years of age on average.)
Having a TET2 mutation seems to predispose myeloid cells to become neoplastic later in life. It is important to distinguish that the TET2 mutation seems to “allow” this transformation rather than causing it. In mice who don’t have the TET2 gene and thus don’t have the TET2 enzyme, stem and progenitor cells have trouble maintaining balance and spontaneously become neoplastic later in life. In TET2 deficient cells, mast cells with D816V mutation show increase in proliferation and survival as opposed to those without with normal TET2 levels. Presence of TET2 in addition to the presence of CKIT D816V mutation is associated with more aggressive forms of SM (including ASM, MCL and SM-AHNMD.)
References:
Damaj, G., Joris, M., Chandersris, O., Hanssens, K., Soucie, E., Canioni, D., et al., 2014.ASXL1 but not TET2 Mutations Adversely Impact Overall Survival of PatientsSuffering Systemic Mastocytosis with Associated Clonal Hematologic Non-Mast-Cell Diseases. PLoS ONE 9 (1), e85362.
Schwaab, J., Schnittger, S., Sotlar, K., Walz, C., Fabarius, A., Pfirrmann, M., et al., 2013.Comprehensive mutational profiling in advanced systemic mastocytosis. Blood122 (October (14)), 2460–2466.
Soucie, E., Hanssens, K., Mercher, T., Georgin-Lavialle, S., Damaj, G., Livideanu, C.,et al., 2012. In aggressive forms of mastocytosis. TET2 loss cooperates with c-KITD816V to transform mast cells. Blood 120 (December (24)), 4846–4849.
Soucie, E., Brenet, F., Dubreuil, P. Molecular basis of mast cell disease. Molecular Immunology 63 (2015) 55-60.